Faster or better? Faster and better?
Multi-dimensional NMR is incredibly powerful, and often for all but the simplest problems; essential for structural elucidation. Sure you can spend time planning, running and interpreting a lot of different 1D experiments that could solve your problem; but often typing a few commands can give you a 2D spectrum that shows you at a glance what you need to see.
Its just a shame it takes so long… A 2D result typically consists of 256 1D experiments; each with x number of scans. If you need to increase the signal to noise; or increase the resolution (in the derived dimensions), the length of time it takes goes up geometrically; its even worse the higher number of dimensions you need to use…
But thinking about just a 2D experiment; if you look at it; its mostly empty space. Really the information you get out of it is list of peaks, described by two frequencies… Surely there ought to be a way to ‘compress’ this at the acquisition level?
Turns out there is; using several different methods. As to how they work; well much the same way as the ‘inertial dampeners’ do on the Star Ship Enterprise; which is to say ‘very well’ I really don’t have the mathematical capability to do a proper explanation. Best I can do is analogies… Which can only go so far, and may actually contravene the laws of physics!
Non-Uniform Sampling.
I asked the question in an early post ‘How many points do you need to represent a sine wave?’ Turns out, not so many… The way this ‘NUS’ technique works is to only sample some of the points in the derived dimensions. For a 2D spectrum; maybe around 25%, for a 3D maybe around 5%; this gives a huge speed up. You then use signal processing techniques to recover the missing points.
So what you do is prepare a ‘sparse sample schedule’ which contains a list of the slices; that is the 1D spectra with the corresponding delays from the full 2D. Ordinarily; you setup your 256 increment HSQC and you get 256 1D spectra each run with equally incremented delays. Using NUS, you might get 32 1D spectra; each run with non-equally incremented delays.
This method works very well for experiments such as HSQC where you tend to only have one peak at a given frequency in F2 (The carbon dimension); and less well for experiments such as COSY or HMBC where you have multiple peaks at a given F2 frequency. And I can’t really get very nice results at all with NOESY. I believe this is because you inherently need more points to represent the more complicated sine/cosine wave you’re trying to calculate to do the final FT on. The quality is also very dependent on the algorithm used for the reconstruction. The results I’ve obtained have all been processed with the same method using Brukers Topspin software. I’ve tried NMRpipe and MDDnmr; and I have some success with HSQC; but just haven’t had the time to put on the crampons and break out the ice-axe to go up the near vertical learning curve for NMRpipe.
Problems with going too quick.
You might think, ‘Thats amazing! I can run everything four times quicker!!!’
As with everything; you don’t get something for nothing…
So. Problems. If you sample at too low a level; you can find the frequencies of the peaks may shift slightly. Also you’ll get more noise in the spectrum. If you extract a row from the 2D you see this noise is ‘non-guassian’ and has the appearance of spikes scattered randomly through the spectrum.
Except it’s not random. It’s dependent on the sparse sample list you used; some schedules may be better than others for a given distribution of frequencies. I tried a list of prime numbers; gave a *very* poor result. Random points are better; random points with a Poisson distribution bias towards the start may be even better and help with poor s/n.
There’s a whole load of papers about schedule generation; but the upshot seems to be there’s no easy way to calculate the best schedule. Also because you don’t necessarily know what peaks you’re going to see; there may be no general way to calculate the ‘best’ schedule.
If your s/n is inherently small; these artefacts can rapidly overwhelm your sample peaks. So sparse, non-uniform sampling may not be appropriate for very dilute samples. As they say, ‘your mileage may vary’.
I find that with simple organic compounds; running a 256 point HSQC at 25% for 2 scans per increment works well; this can be done in around 2 minutes. You might be able to to it in 1 minute; but I prefer being more certain it will work. For simple compounds, COSY I do at 37.5% of 256, 2 scans; HMBC, 37.5% of 360 and 2 scans. For more resolution I do COSY at 37.5% of 512, HSQC 25% of 1024 and HMBC at 37.5% of 768. You should look at your results critically though, if you’re missing peaks you might expect, further investigation may be required!
Enhance! Enhance!
So you might be able to to things faster; how about better? again, take an HSQC, our standard experiment is 256 increments over about 180ppm; this gives a resolution at 500 MHz of around 180Hz in F1. So the peaks quite often overlap in crowded areas. What can NUS do for this?
Turns out quite a lot.
Here’s a DEPT135 edited HSQC, 25% of 256 points. Took about 2mins 19 seconds. Looks perfectly good…

Here’s a zoom.

Here’s a zoom of 25% of 1024 points; took about 8mins 28 seconds. Looks good…

Lets enhance… 25% of 2048, took less than 17 mins.

Enhance! Enhance! 25% of 24K points! 3hrs 17 mins. A long time; but look at the resolution!

Faster! Faster! The s/n is good; lets do 3.125% of 24K points… Only takes 25 mins. You’ve got to look carefully to see a difference. Note the full 24K points would take 16hrs 37mins…

. I would say the 2D spectra of the 25% version looks slightly better though when you only scale to remove the noise. If you actually look carefully at the noise; the 3.125% (rd) is a lot worse… Its hard to quantify, as its non-Gaussian; but there’s a lot more spikes.

Here’s the calculated sum of both 24K spectra; to produce a calculated DEPT135. Not much noise in either of them

Zoom and enhance… You can resolve peaks about 3Hz apart.


Just to show the same region at 2048K and 256K, where we started.


So, with a nice sample you can run samples faster, even faster and with better resolution in some instances.
APSY
This is another method of speeding up multi-dimensional acquisition of 3 and above.
I think a good way to describe this is to imagine dropping some stones into a pool at the same time… The ripples spread outwards. Then imagine you freeze the surface before the waves hit the edges. If you then take slices through the surface at different angles; how many slices to you need to re-construct the positions of the stones?
I’ve never run any of these; so I can’t say much more about how well it works in practice. About as well as those inertial dampers I expect.
Here’s a reference.
https://www.ncbi.nlm.nih.gov/pubmed/21710379

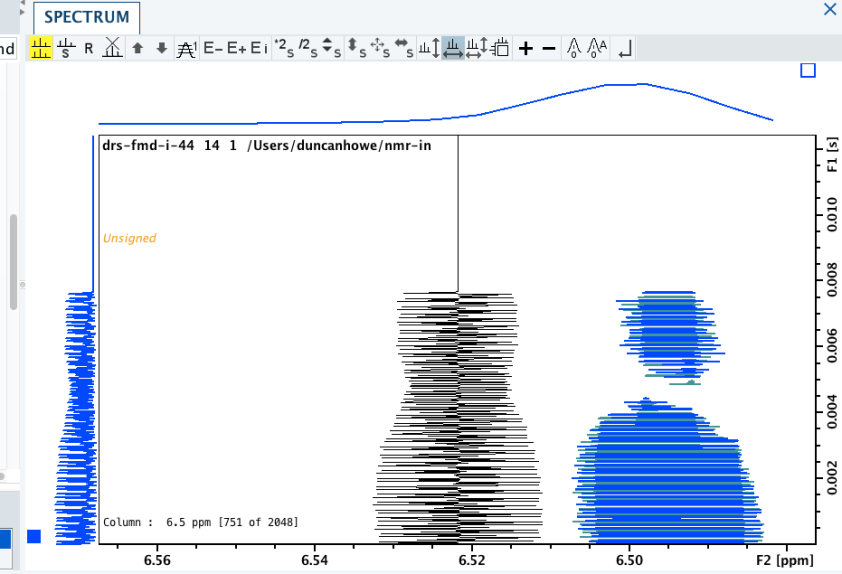 It can be worth looking at the actual rows themselves, the 1d slices that the full experiment is made up of; as you may be able to identify experimental artefacts, which could affect the final result.
It can be worth looking at the actual rows themselves, the 1d slices that the full experiment is made up of; as you may be able to identify experimental artefacts, which could affect the final result.











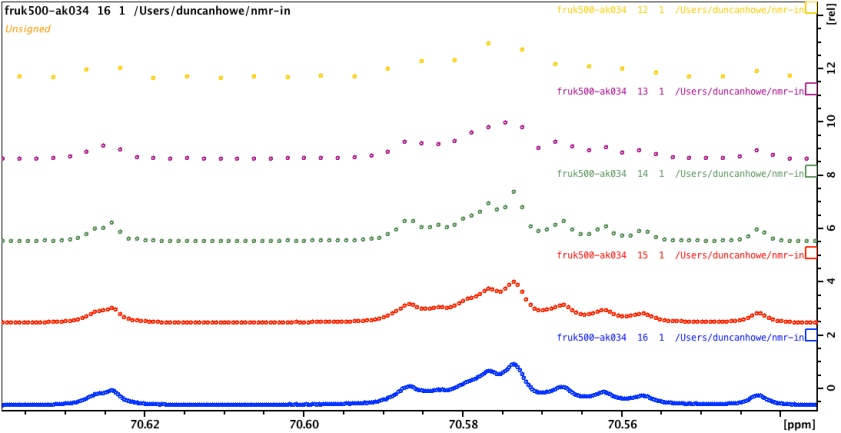






 Zooming into the plot can show you some interesting things… This zoom shows that the temperature of the sample never quite reached perfect stability. This experiment ran a scan every 4 seconds and the temperature ramped up to the next one, 600 seconds after reaching +/- 0.1C stability.
Zooming into the plot can show you some interesting things… This zoom shows that the temperature of the sample never quite reached perfect stability. This experiment ran a scan every 4 seconds and the temperature ramped up to the next one, 600 seconds after reaching +/- 0.1C stability.